Int J Mol Sci. 2013 iulie; 14(7): 14575–14593.
doi: 10.3390/ijms140714575 PMCID: PMC3742260 PMID: 23857055
Li Lin , 1, 2, † Qiong-Xia Huang , 3, † Shu-Sheng Yang , 2 Jiang Chu , 1 Jian-Zhi Wang , 1, * și
Qing Tian 1, *
Informații despre autor Note despre articol Informații privind drepturile de autor și licență Declinare a răspunderii
Mergi la:
Abstract
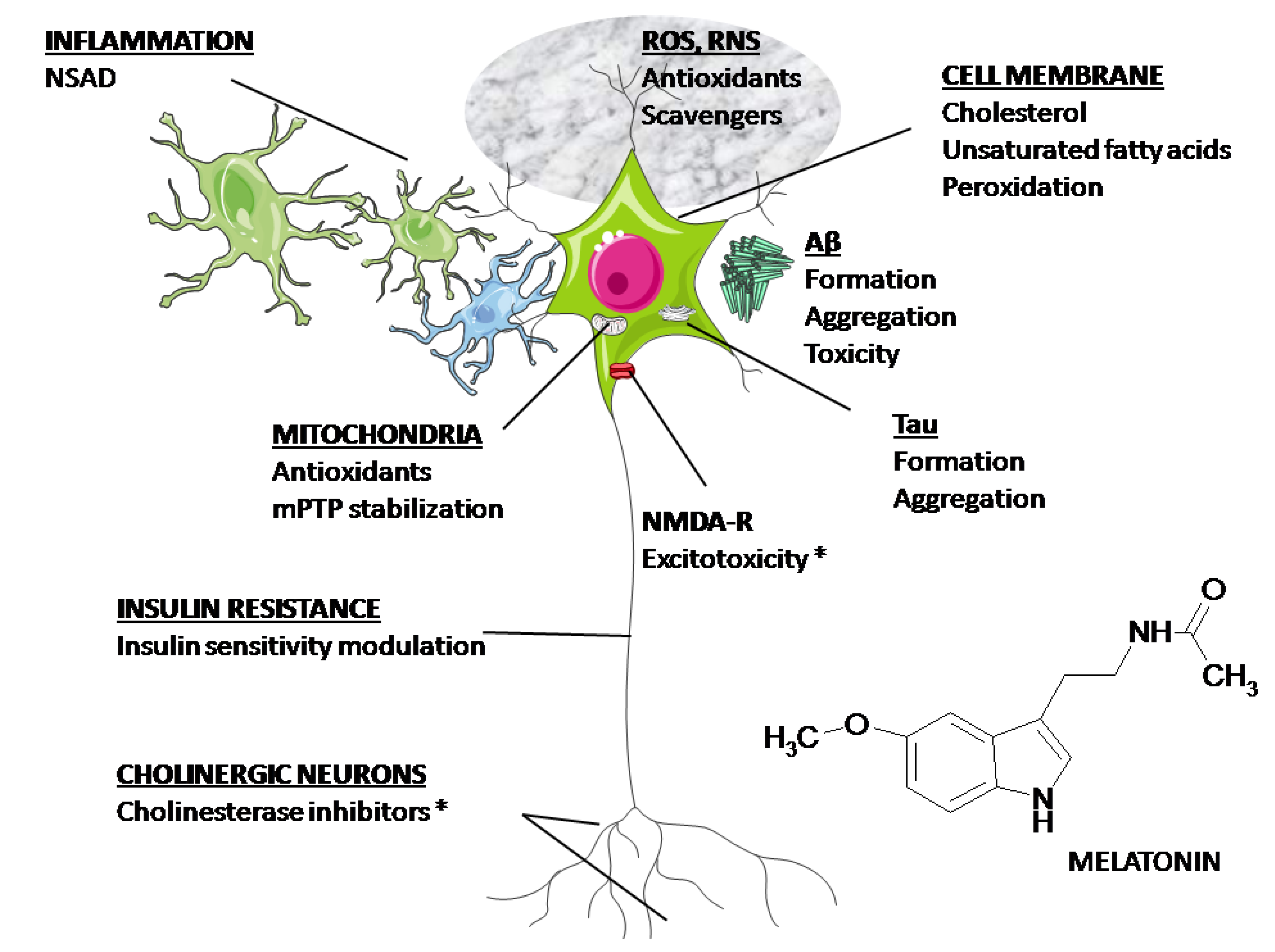
Boala Alzheimer (AD), o tulburare neurodegenerativă legată de vârstă, cu deficit de cogniție progresiv, este caracterizată prin plăci senile extracelulare (SP) de β-amiloid (Aβ) agregat și încurcături neurofibrilare intracelulare, care conțin în principal proteina tau hiperfosforilată asociată microtubulilor. Factori multipli contribuie la etiologia AD în ceea ce privește inițierea și progresia. Melatonina este un hormon produs endogen în creier și scade în timpul îmbătrânirii și la pacienții cu AD. Datele din studiile clinice indică faptul că suplimentarea cu melatonină îmbunătățește somnul, ameliorează apusul soarelui și încetinește progresia deteriorării cognitive la pacienții cu AD. Melatonina protejează eficient celulele neuronale de toxicitatea mediată de Aβ prin proprietăți antioxidante și anti-amiloide. Nu numai că inhibă generarea de Aβ, dar oprește și formarea fibrilelor de amiloid printr-o interacțiune dependentă de structură cu Aβ. Studiile noastre au demonstrat că melatonina atenuează eficient hiperfosforilarea tau asemănătoare cu Alzheimer. Deși mecanismul exact nu este încă pe deplin înțeles, se propune o influență reglatoare directă a melatoninei asupra activităților protein kinazelor și proteinelor fosfataze. În plus, melatonina joacă, de asemenea, un rol în protejarea sistemului colinergic și în antiinflamație. Scopul acestei revizuiri este de a stimula interesul pentru melatonină ca agent potențial util în prevenirea și tratamentul AD.
1. Introducere
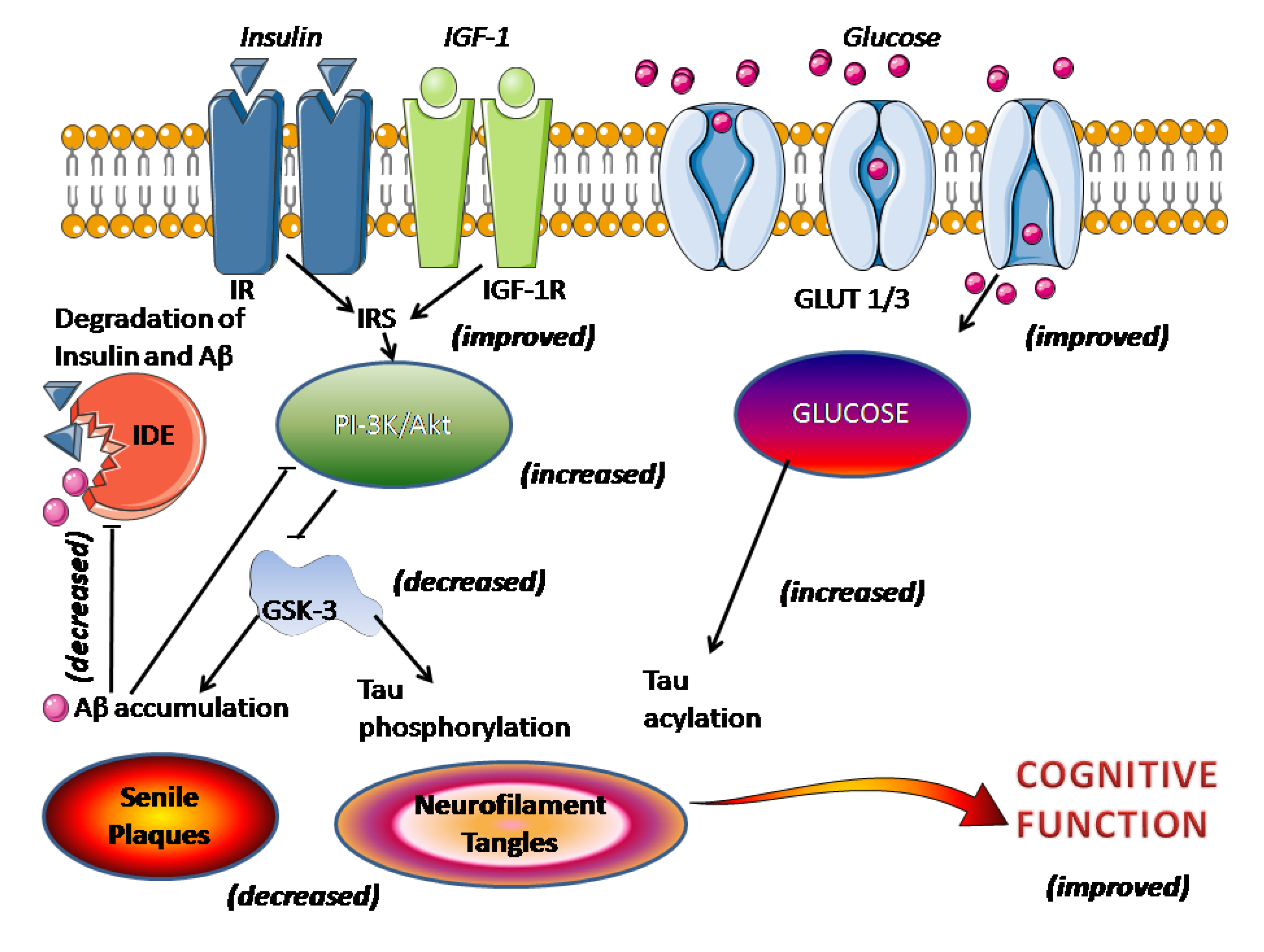
Boala Alzheimer (AD) este o boală neurodegenerativă asociată vârstei și caracterizată prin pierderea progresivă a cogniției și alte manifestări neurocomportamentale. Semnele patologice ale AD includ plăci senile extracelulare (SP), constând în principal din β-amiloid (Aβ) și încurcături neurofibrilare intracelulare (NFT), compuse în principal din tau hiperfosforilat anormal, o proteină asociată cu microtubuli [1 ] . În ciuda unui număr mare de studii întreprinse, etiologia AD este în mare parte necunoscută. Au fost propuse multe mecanisme, inclusiv predispoziții genetice (de exemplu, niveluri de expresie și subforme ale presenilinelor (PS) și apolipoproteinei (Apo) E), procese inflamatorii asociate cu eliberarea de citokine, stres oxidativ și neurotoxicitate de către ionii metalici [ 2 – 6 ] .].
Melatonina ( N -acetil-5-metoxitriptamina), un metabolit al triptofanului și sintetizat în principal în glanda pineală, are o serie de funcții fiziologice, inclusiv reglarea ritmurilor circadiene, curățarea radicalilor liberi, îmbunătățirea imunității și, în general, inhibarea oxidării biomoleculelor. Scăderea melatoninei în ser și lichidul cefalorahidian (LCR) și pierderea ritmului diurn al melatoninei sunt observate la pacienții cu AD [ 7 – 12 ]. Mai mult, nivelul melatoninei din LCR scade odată cu progresia neuropatologiei AD, așa cum este determinat de etapele Braak [ 12] .]. Nivelurile melatoninei atât în LCR, cât și în glanda pineală umană postmortem sunt deja reduse la subiecții cu AD preclinici, care sunt încă intacte din punct de vedere cognitiv și au doar cele mai timpurii semne de neuropatologie AD [8 , 12 ] . Există o corelație puternică între conținutul pineal și nivelul melatoninei LCR [ 8 ] și între LCR și nivelurile de melatonină plasmatică [ 7 ], sugerând că un nivel redus de melatonină LCR poate servi ca un marker timpuriu pentru primele etape ale AD. La mamifere, melatonina își exercită unele dintre funcțiile sale prin doi receptori specifici de membrană cu afinitate mare, receptorul 1 al melatoninei (MT1) și receptorul 2 al melatoninei (MT2). Scăderea imunoreactivitatii MT2 și imunoreactivitatea MT1 crescută au fost raportate în hipocampul pacienților cu AD.13 , 14 ]. Deși glanda pineală a pacienților cu AD are modificări moleculare, nu s-au observat modificări ale greutății pineale, calcifierii sau conținutului total de proteine [ 8 , 15 ]. De asemenea, se arată că ARNm a receptorului β1-adrenergic a dispărut, iar activitatea și expresia genică a monoaminoxidazei (MAO) au fost reglate în sens pozitiv la pacienții cu AD, sugerând că dereglarea inervațiilor noradrenergice și epuizarea serotoninei, precursorul melatoninei, ar putea fi responsabil pentru pierderea ritmului melatoninei și reducerea nivelului de melatonina în AD [ 16]. Suplimentarea cu melatonină a fost sugerată pentru a îmbunătăți ritmicitatea circadiană, de exemplu, scăderea comportamentului agitat, a confuziei și a „apusului soarelui” și pentru a produce efecte benefice asupra memoriei la pacienții cu AD [17-21 ] . Prin urmare, suplimentarea cu melatonină, cu toxicitatea sa scăzută marcată [ 22 – 24 ], poate fi una dintre posibilele strategii pentru tratamentul simptomatic.
În AD, se crede, în general, că Aβ joacă un rol important în promovarea degenerării neuronale, făcând neuronii mai vulnerabili la creșterile legate de vârstă ale nivelurilor de stres oxidativ și deteriorări în metabolismul energetic celular [25 ] . Ca proteină principală asociată cu microtubuli, tau promovează asamblarea microtubulilor și stabilizează microtubulii. Hiperfosforilarea va reduce în mod evident abilitățile tau, ceea ce duce la perturbarea aranjamentului citoscheletic [ 26 , 27 ]. Amploarea patologiei neurofibrilare, și în special numărul de NFT cortical, se corelează pozitiv cu severitatea demenței [ 28] .]. Deoarece melatonina este capabilă să îmbunătățească unele dintre simptomele clinice ale AD, iar nivelul melatoninei scade dramatic în timpul AD, studiile privind relația dintre melatonină și patologia AD vor fi utile pentru a evalua potențialul său în prevenirea sau tratamentul AD. În această revizuire, vom aborda rolul melatoninei în hiperfosforilarea tau și toxicitatea Aβ. Deoarece deficitul colinergic și inflamația sunt implicate în patogenia AD, se introduce și protecția melatoninei asupra sistemului colinergic și a inflamației. Fiecare parte este descrisă, de la observarea fenomenului până la investigarea mecanismelor și speculații.
Mergi la:
2. Melatonina în hiperfosforilarea Tau
Tau hiperfosforilat a fost identificat în mai mult de o duzină de tulburări neurodegenerative, denumite tauopatii, inclusiv AD, boala Niemann-Pick de tip C și așa mai departe [ 29-31 ]. Dintre aceste tulburări legate de tau, AD este cea mai frecventă și cea mai bine studiată tauopatie. În creierul AD, nivelul tau-ului hiperfosforilat este de 3-4 ori mai mare decât cel al tau-ului din creierul adult normal [ 32 , 33] .]. Există 79 de situsuri presupuse de fosforilare a serinei sau treoninei în cea mai lungă izoformă tau a creierului uman și mai mult de 30 de situsuri de fosforilare au fost identificate în creierul AD, inclusiv Thr39, Ser46, Thr50, Thr69, Thr153, Thr175, Thr181, Ser184, Ser185. , Ser198, Ser199, Ser202, Thr205, Ser208, Ser210, Thr212, Ser214, Thr217, Thr231, Ser235, Ser237, Ser238, Ser245, Ser258, Ser262, Ser285, Ser262, Ser285, Ser293, Ser3, Ser3, Ser30, Ser293, Ser3, Ser3, Ser35 r377, Ser396 , Ser400, Thr403, Ser404, Ser409, Ser412, Ser413, Ser416 şi Ser422 [ 34 – 39 ]. Răspândirea patologiei tau în creier este semnul distinctiv al patogenezei AD, iar numărul de NFT este corelat pozitiv cu deficitul cognitiv clinic al pacienților cu AD [ 40 ].
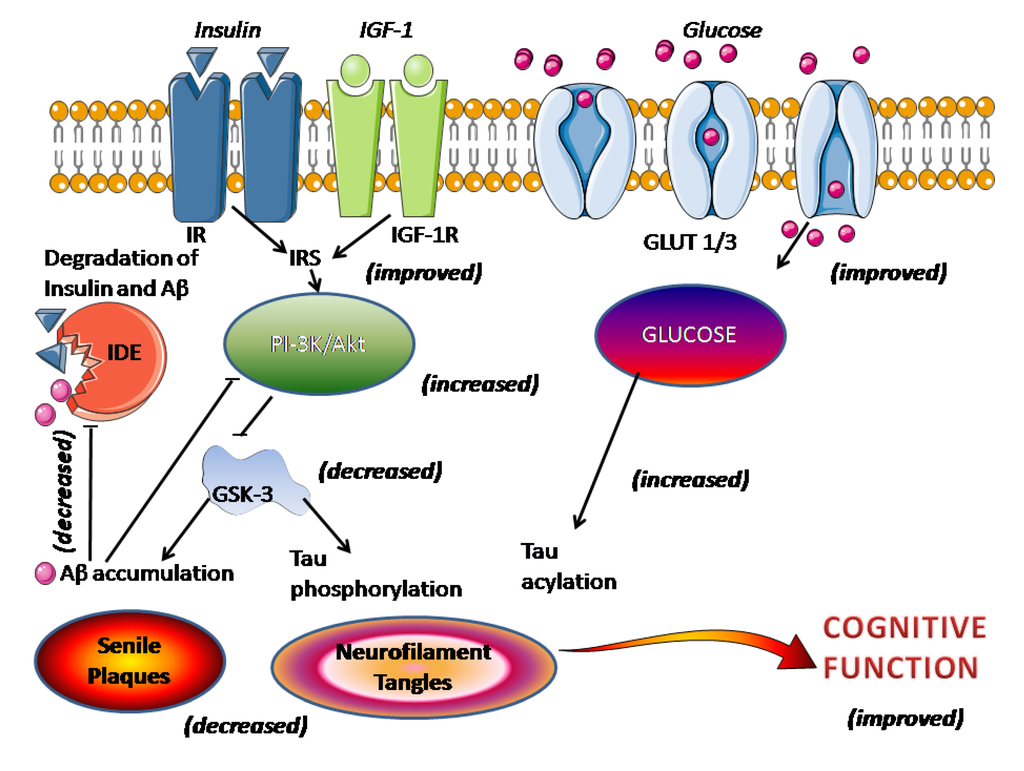
Inhibarea hiperfosforilării tau este o țintă în tratamentul AD. Astfel, am studiat sistemic efectul melatoninei asupra hiperfosforilării tau și am constatat că melatonina atenuează eficient hiperfosforilarea tau indusă de wortmannin [ 41 ], calyculin A (CA) [ 42 – 44 ] și acid okadaic [ 45 ] în neuroblastom N2a și SH-SY5Y. celule. S-a demonstrat în continuare că melatonina a ameliorat semnificativ hiperfosforilarea tau provocată de wortmannin [ 46 ], izoproterenol [ 47 , 48 ], CA [ 44 ] și iluminarea constantă a luminii [ 24]] la șobolani. Pentru a elucida mecanismele care stau la baza efectului inhibitor al melatoninei asupra hiperfosforilării tau, au fost detectate modificări ale activităților protein kinazelor și fosfatazelor. Tratamentul cu melatonină nu numai că a inhibat activarea glicogen sintazei kinazei-3 (GSK-3) indusă de wortmannin, activarea proteinei kinazei A (PKA) indusă de izoproterenol și inactivarea proteinei fosfatazei-2A (PP-2A) indusă de CA, dar a antagonizat și oxidarea. stresul indus de acești agenți [ 46 , 49 , 50 ]. Aceste rezultate din studiile noastre oferă dovezi pentru eficacitatea puternică a melatoninei în inhibarea hiperfosforilării tau.
Pentru a explora dacă o scădere a melatoninei ar induce alterarea fosforilării tau, am inhibat biosinteza melatoninei prin injectarea creierului de haloperidol, un inhibitor al 5-hidroxiindol-O-metiltransferazei (o enzimă cheie în sinteza melatoninei) la șobolani [ 51 ]. S-a descoperit că inhibarea biosintezei melatoninei nu numai că a dus la afectarea memoriei spațiale la șobolani, dar a indus și o creștere a fosforilării tau cu o scădere concomitentă a activității PP-2A. Suplimentarea cu melatonină prin injectare prealabilă timp de o săptămână și întărirea în timpul perioadei de administrare a haloperidolului au îmbunătățit semnificativ deficitele de retenție a memoriei, au oprit hiperfosforilarea tau și stresul oxidativ și au restabilit activitatea PP-2A [51] .]. De asemenea, am folosit iluminare constantă pentru a întrerupe metabolismul melatoninei la șobolani. Concomitent cu scăderea melatoninei serice, șobolanii luminați în mod constant au dezvoltat deficite de memorie spațială, hiperfosforilarea tau la mai multe locuri, activarea GSK-3 și PKA, precum și suprimarea PP-1. Leziuni oxidative proeminente și leziuni de organele, demonstrate prin exprimarea crescută a proteinelor legate de stresul reticulului endoplasmatic (ER), inclusiv proteina de legare a imunoglobulinei (BiP)/GRP78 și CHOP/GADD153, au scăzut numărul de ER rugoase și ribozomi liberi, au dus la mai subțire. sinapsele și creșterea superoxid dismutazei și MAO, care au fost observate și la șobolanii expuși la lumină. Suplimentarea simultană cu melatonină a oprit parțial deficiențele comportamentale și moleculare [ 24]. Deși nu este clar dacă concentrația scăzută de melatonina este un factor cauzal în patologia AD sau doar un proces secundar, rezultatele noastre implică puternic rolul important al scăderii melatoninei în afectarea memoriei spațiale asemănătoare Alzheimer și hiperfosforilarea tau.
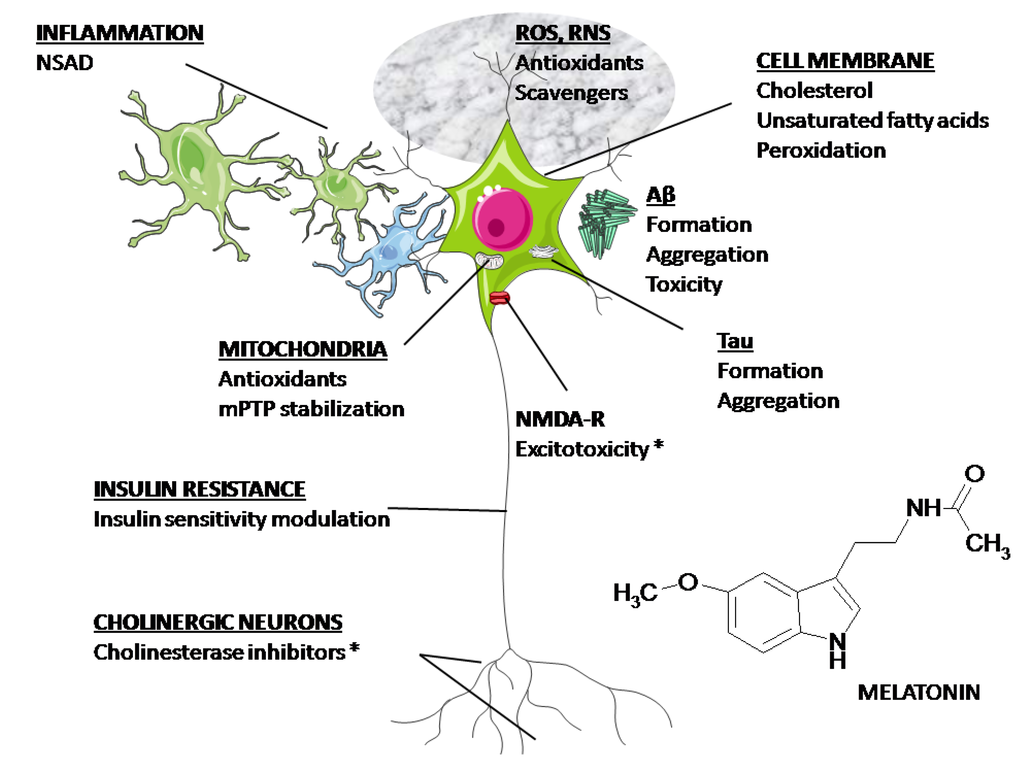
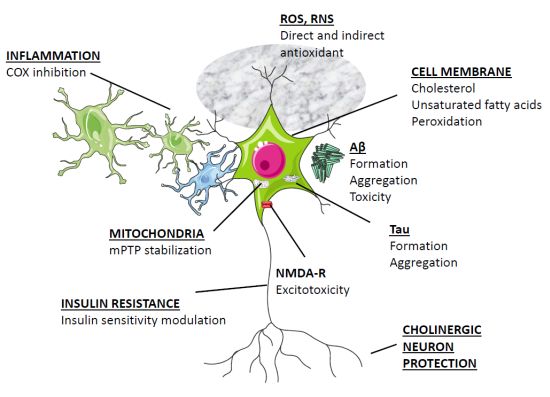
Agenții chimici utilizați în studiile noastre, inclusiv wortmannin, izoproterenol și CA, nu numai că au indus fosforilarea tau, dar au inițiat și stresul oxidativ, manifestat printr – un nivel crescut de malondialdehidă și o activitate alterată a superoxid dismutază (SOD) [41-43 ] . În plus, melatonina este un puternic captator direct de radicali liberi și un antioxidant indirect care acționează prin creșterea activității mai multor enzime antioxidante importante, cum ar fi SOD, glutation peroxidaza și glutation reductază [52 ] . Iluminarea constantă nu numai că a indus scăderea nivelului de melatonină seric, ci și nivelurile crescute de SOD și MAO la șobolani [ 24 ]. Se știe că stresul oxidativ influențează starea de fosforilare a tau [ 53– 55 ]. Acumularea de proteine greșite și agregate în neuronii creierului AD a fost considerată a fi legată de stresul oxidativ, împreună cu modificările structurii sale moleculare odată cu îmbătrânirea [ 56 ]. Ca antioxidant și captator de radicali liberi [ 57 – 59 ], melatonina previne supraproducția de radicali liberi și reduce daunele neuronale rezultate dintr-o varietate de procese patologice [ 60 – 62 ]. Prin urmare, este posibil ca prevenirea fosforilării tau de către melatonină să se datoreze parțial activității sale antioxidante.
Unele studii au indicat, de asemenea, că melatonina poate acționa ca un modulator al enzimei într-un mod care nu are legătură cu proprietățile sale antioxidante. Datele acumulate oferă dovezi pentru reglarea de către melatonină a protein kinazelor, inclusiv PKA [ 63 , 64 ], protein kinaza C (PKC) [ 65 , 66 ], Ca 2+ /calmodulin-dependent kinaze II (CaMKII) [ 67 , 68 ] și familia protein kinazei activate de mitogen (MAPK) [ 69 – 72 ]. Corelația documentată între melatonină și AMPc indică faptul că melatonina ar putea inhiba activitatea PKA prin inhibarea cuplată a receptorului de melatonină a adenilil ciclazei și reducerea AMPc [ 63 ]., 64 ]. Deși nu există dovezi ale unei relații directe între melatonină și activitatea GSK-3, unul dintre studiile noastre a constatat că tratamentul cu melatonină a revizuit iluminarea constantă a luminii indusă activarea GSK-3 în creierul șobolanilor [24 ] .
Mergi la:
3. Melatonina și toxicitatea Ap
Aβ, care compune 39-43 de resturi de aminoacizi derivate din precursorul său, proteina precursor de amiloid (APP), joacă un rol esențial în patogeneza AD [ 25 , 73 ]. APP matur este procesat proteolitic prin căi distincte de α-secretază sau β-secretază [ 74 ]. Calea α non-amiloidogenă implică scindarea APP în secvența Ap de către α-secretază pentru a elibera un fragment APP N-terminal, care, la rândul său, este scindat de y-secretază. Astfel, scindarea de către y-secretază exclude formarea Ap. Calea β-secretazei amiloidogenă, care are ca rezultat formarea peptidei Aβ intacte, este mediată de scindarea secvenţială a β-secretazei şi γ-secretazei la nivelul N- şi C.-terminale ale secvenței Aβ, respectiv [ 75 ]. S-a descoperit că melatonina inhibă nivelurile normale de secreție de APP solubilă (sAPP) în diferite linii celulare prin interferarea cu maturarea completă a APP [ 76 ]. În plus, administrarea melatoninei a redus eficient generarea și depunerea Aβ atât in vivo [ 77 , 78 ], cât și in vitro [ 76 , 79 – 81 ]. Am demonstrat că melatonina reduce generarea de Ap în celulele N2a de neuroblastom de șoarece care adăpostesc APP695 [ 80 , 82 ].
Cu toate acestea, un studiu in vivo a arătat că melatonina nu a afectat expresia holoproteinei APP la șoarecii transgenici Tg2576 [ 77 ]. Mai mult, în ciuda atingerii unor concentrații plasmatice mari de melatonină, terapia cronică cu melatonină la șoarecii bătrâni Tg2576 inițiată la vârsta de 14 luni nu numai că nu a reușit să îndepărteze plăcile existente, dar nu a reușit să prevină depunerea suplimentară de Aβ [83 ] . Acest rezultat este în contrast cu cele ale scăderii Ap la șoarecii de tip sălbatic tratați cu melatonină [ 78 ] și nitrarea redusă a Ap și proteinei la șoarecii Tg2576 tratați cu melatonină [ 77 ]. Timpul de inițiere a tratamentului cu melatonină ar putea explica diferența dintre studiile lui Matsubara și colab. [ 77] și Quinn și colab. [ 83 ], în care a fost utilizat același model de șoarece transgenic Tg2576. Patologia plăcii de amiloid apare de obicei la șoarecii Tg2576 de 10-12 luni [ 84 ]. Tratamentul cu melatonină în studiul lui Matsubara și colab. a fost început când șoarecii aveau patru luni (înainte de apariția plăcilor hipocampale și corticale) [ 77 ], o etapă patologică mai timpurie comparativ cu vârsta de 14 luni în studiul lui Quinn și colab. [ 83]. Cu toate acestea, ambele studii sunt de acord în a găsi puține dovezi ale efectelor antioxidante puternice ale melatoninei la cei mai bătrâni șoareci. Aceste descoperiri indică faptul că melatonina are capacitatea de a regla metabolismul APP și de a preveni patologia Ap, dar nu reușește să exercite efecte anti-amiloid sau antioxidante atunci când este inițiată după depunerea Ap. Deși s-au obținut concluzii consistente, niciunul dintre studiile înrudite nu explică în continuare modul în care melatonina își exercită efectul inhibitor asupra generării Ap. Scindarea proteolitică a APP de către calea α-secretazei este reglată de mulți stimuli fiziologici și patologici, iar mecanismul dependent de PKC este unul dintre cei mai recunoscuti. Stimulii, cum ar fi agoniştii muscarinici şi metabotrofici ai receptorilor de glutamat şi esterii de forbol, au capacitatea de a stimula secreţia de APP solubilă şi de a inhiba formarea de Aβ prin activarea PKC.75 ]. Mecanismul prin care activitatea PKC crește secreția de APP solubilă este încă necunoscut, dar poate implica pași adiționali de kinază și eventuala activare a secretazelor care au mediat clivajul APP. Recent, reglarea inhibitorie prin inhibarea GSK-3 asupra generării Aβ a fost bine stabilită [ 85-87 ]. Mecanismul din spatele acestui lucru nu este clar. S-a demonstrat că inhibarea GSK-3 și reglarea în sus a kinazei N -terminale c-Jun (JNK) au ca rezultat o activitate crescută a metaloproteazei matricei și o degradare crescută a Ap [ 88 ]. Deoarece fosforilarea GSK-3 duce la inactivarea sa, datele sugerează că GSK-3 activat poate inhiba sau reduce activarea JNK de către anumiți stimuli [ 89] .]. GSK-3 interacționează cu presenilin-1 (PS1), un cofactor pentru y-secretaza, ceea ce implică faptul că GSK-3 poate funcționa ca o componentă în complexul y-secretază [90 , 91 ] . Presupunând că melatonina poate influența activitatea PKC și GSK-3 așa cum sa menționat mai devreme, se postulează că melatonina poate regla procesarea APP prin căile PKC și GSK-3. Deoarece PKC este un regulator în amonte al GSK-3, GSK-3 poate fi una dintre căile de semnal obișnuite care cresc generarea de Ap și hiperfosforilarea tau. Reglarea formării fibrilelor Aβ și proprietățile patologice importante ale Apei, cum ar fi neurotoxicitatea și rezistența la degradarea proteolitică, depind de capacitatea peptidelor de a forma structuri β-sheet și/sau fibrile de amiloid [92 , 93 ] .
Intervenția în procesul de agregare Aβ poate fi considerată o abordare pentru oprirea sau încetinirea progresiei AD. Melatonina poate interacționa cu Aβ40 și Aβ42 și poate inhiba formarea progresivă a fibrilelor β-sheet și/sau amiloid [ 94 – 96 ]. Efectul anti-fibrilogenic al melatoninei a fost demonstrat prin diferite tehnici, inclusiv spectroscopie circulară cu dicroism (CD), microscopie electronică și spectroscopie prin rezonanță magnetică nucleară (RMN) și spectrometrie de masă cu ionizare prin electrospray (ESI-MS) [95 ] . Mai mult, interacțiunea dintre melatonină și Aβ pare să depindă de caracteristicile structurale ale melatoninei, mai degrabă decât de proprietățile sale antioxidante, deoarece nu ar putea fi reprodusă de analogii melatoninei sau de alți captatori de radicali liberi.92 , 94 ]. Dovezile derivate din ESI-MS au demonstrat că a existat o interacțiune hidrofobă între Ap și melatonină, iar investigațiile proteolitice au sugerat că interacțiunea a avut loc pe cele 29-40 de reziduuri ale segmentului Aβ [95 ] . Rezultatele spectroscopiei RMN au confirmat în continuare o interacțiune specifică reziduului între melatonină și oricare dintre cele trei reziduuri de histidină și aspartat ale Ap [ 96 ]. Punțile de sare imidazol-carboxilat formate de lanțurile laterale ale reziduurilor de histidină (His+) și aspartat (Asp−) sunt esențiale pentru formarea structurilor de foi de amiloid β [97], iar ruperea acestor punți de sare promovează dizolvarea fibrilei [ 98 ] ].
Melatonina ar putea promova conversia foilor β în bobine aleatorii prin întreruperea punților de sare imidazol-carboxilat și, astfel, să prevină fibrilogeneza și agregarea Ap. Prin urmare, este posibil ca prin blocarea formării conformației secundare a foii p, melatonina nu numai că poate reduce neurotoxicitatea, ci și să faciliteze clearance-ul peptidei prin degradarea proteolitică crescută. S-a demonstrat că melatonina interacționează direct cu Aβ și previne agregarea acestuia [ 99 ]. Tratamentul cu Aβ provoacă un spectru de leziuni celulare, inclusiv creșterea peroxidării lipidelor și a concentrației de calciu liber intracelular, deteriorarea oxidativă a ADN-ului mitocondrial și apariția markerilor apoptotici [100] .]. Mitocondriile nu sunt doar locul principal de generare a speciilor reactive de oxigen (ROS), ci și ținta principală de atac pentru ROS. S-a considerat că melatonina stabilizează fluiditatea membranelor interne mitocondriale; iar legarea de membranele mitocondriale a fost dezvăluită [ 101]. Stresul oxidativ acționează sinergic cu perturbarea homeostaziei calciului intracelular. Deteriorarea membranei indusă de radicalii liberi induce un aflux suplimentar de calciu, iar influxul accentuat de calciu rezultat, la rândul său, va induce generarea de alți radicali liberi. Prin urmare, stresul oxidativ joacă un rol central în neurotoxicitatea indusă de Ap și chiar în moartea celulelor. Pe lângă faptul că Aβ provoacă stres oxidativ, s-a propus că daunele oxidative ar putea exacerba un cerc vicios, în care procesarea amiloidogenă a APP ar fi facilitată și mai mult pentru a genera mai mult Ap, care, la rândul său, crește stresul oxidativ [102 ] . Acumularea datelor implică faptul că melatonina protejează eficient celulele împotriva daunelor oxidative induse de Aβ și a morții celulare in vitro [ 103 ,104 ] şi in vivo [ 77 , 105 – 107 ]. În celulele și animalele tratate cu Aβ, melatonina își exercită activitatea de protecție în principal printr-un efect antioxidant, în timp ce în celulele transfectate cu APP și modelele animale transgenice, mecanismul de bază este atribuit nu numai proprietății sale antioxidante, ci și proprietății sale anti-amiloid. incluzând inhibarea atât a generării de Ap cât și a formării de foi de p și/sau fibrile de amiloid.
În plus, unele descoperiri sugerează un rol pentru semnalizarea perturbată a melatoninei în tulburările de somn care sunt comune la pacienții cu AD [ 108-110 ]. Prin microdializă in vivo la șoarecii Tg2576, s-a constatat că cantitatea de lichid interstițial cerebral (ISF) Ap a corelat cu starea de veghe. În plus, ISF Aβ a crescut semnificativ și în timpul privării acute de somn, dar a scăzut cu perfuzia unui antagonist dublu al receptorului de orexină. Restricția cronică a somnului a crescut semnificativ formarea plăcii Aβ [ 111 ]. Mai mult, administrarea melatoninei a redus eficient generarea și depunerea de Aβ, atât in vivo , cât și in vitro [ 76 – 81] .]. Astfel, melatonina poate inhiba generarea și încărcarea Ap, dar mecanismul necesită investigații suplimentare.
Mergi la:
4. Protecția Melatoninei asupra sistemului colinergic
Deficitul sistemului colinergic este, de asemenea, un eveniment precoce și primar în patogeneza AD [ 112 ]. Neuronii din nucleul bazal al lui Meynert, o sursă majoră de inervație colinergică a cortexului cerebral și a hipocampului, suferă o degenerare profundă și selectivă în creierul AD [ 113 – 115 ]. Nivelul de acetilcolină (ACh) este scăzut în stadiul incipient al AD, în timp ce activitățile enzimei de sinteză, colin acetiltransferazei (ChAT) și enzimei de hidrolizare, acetilcolinesteraza (AChE), nu se modifică până în stadiul târziu al AD. 116 – 118]. Alte investigații biologice ale țesutului din biopsie și autopsie au găsit o scădere profundă a activității ChAT în neocortexul pacienților cu AD, corelând pozitiv cu severitatea demenței [ 102 ]. Deși mecanismul care duce la deficitul de ACh este încă necunoscut, inhibitorul AChE a fost folosit ca tratament și este considerat standardul de îngrijire pentru tratamentul pentru AD ușoară până la moderată [119 ] .
Melatonina are efecte protectoare asupra sistemului colinergic. Un studiu anterior a demonstrat, de asemenea, că melatonina a prevenit parțial inhibarea indusă de peroxinitrit a transportului de colină și a activității ChAT în mai multe proteine neuronale din sinaptozomi și, mai ușor, din veziculele sinaptice [120 ] . În plus, se raportează că tratamentul de patru luni cu melatonină a ameliorat semnificativ modificările neuropatologice, comportamentale și biochimice la șoarecii transgenici APP695 de opt luni cu depunere de Aβ, deficit semnificativ de învățare și memorie și o reducere profundă a activității ChAT în cortexul frontal și hipocampus [ 105]. Un alt studiu a mai arătat că un tratament similar cu melatonina a antagonizat deficitul de memorie spațială și a scăzut activitatea ChAT la șobolanii adulți ovariectomizați [ 121 ]. Cu toate acestea, la șobolanii infuzați intracerebroventricular cu Aβ timp de 14 zile, în care activitatea ChAT a fost redusă semnificativ, melatonina nu a putut restabili activitatea acestei enzime [ 122 ]. Melatonina a arătat inhibarea numai a creșterii induse de lipopolizaharide (LPS) a activității AChE, în timp ce nu au fost observate modificări la șobolanii tratați cu LCR. Aceste rezultate sunt în sprijinul influenței inhibitorii a melatoninei asupra activității AChE în demența indusă de streptozotocină [ 123 ].
În comparație cu placebo, inhibitorii de colinesterază, cum ar fi donepezilul, tacrina, rivastigmina și galantamina, care pot rezolva deprivarea de acetilcolină, sunt capabili să stabilizeze sau să încetinească declinul cogniției, funcției, comportamentului și schimbării globale [124] .]. Secreția de melatonină scade în AD, iar această scădere a fost postulată ca fiind responsabilă pentru dezorganizarea circadiană, scăderea eficienței somnului și afectarea funcției cognitive observate la acești pacienți. Înlocuirea melatoninei s-a dovedit eficientă în tratarea apusului soarelui, a deficienței cognitive ușoare (MCI), un sindrom etiologic eterogen care precede demența și alte tulburări de veghe în somn la pacienții cu AD. Pe lângă inhibarea activității AChE, perspectivele pentru melatonina ca tratament pentru AD se bazează, de asemenea, pe eliminarea speciilor reactive de oxigen și azot, rezolvarea tulburărilor de somn, scăderea toxicității Aβ și a încărcării [125] .]. Nu există dovezi clinice care să arate care este mai bun pentru pacienții cu AD în ceea ce privește inhibitorul AChE și melatonina. Cu toate acestea, combinația dintre aceste două medicamente poate avea efecte mult mai bune. Recent, hibrizii tacrină-melatonină au fost proiectați și sintetizați ca noi candidați multifuncționali pentru AD [ 126 , 127 ]. Compușii prezintă proprietăți colinergice și antioxidante îmbunătățite, fiind inhibitori mai puternici și selectivi ai AChE uman decât tacrina și captând radicalii liberi mai bine decât melatonina. Ele prezintă o toxicitate scăzută și pot fi capabile să pătrundă în sistemul nervos central [ 126 ]. Administrarea directă intracerebrală a unuia dintre acești hibrizi, N-(2-(1H-indol-3-il)etil)-7-(1,2,3,4-tetrahidroacridin-9-ilamino) heptanamidă, scăderea morții celulare induse de Ap și a încărcăturii cu amiloid în parenchimul cerebral al APP /Soareci PS1. Mai mult, reducerea patologiei Aβ a fost însoțită de o recuperare a funcției cognitive [ 127 ].
Mergi la:
5. Rolul melatoninei în neuroinflamația AD
Un factor comun în patogenia AD este supraactivarea microgliei cu supraexpresia ulterioară a citokinelor proinflamatorii [ 128 – 130 ]. Acumularea de Aβ în plăci, precum și oligomerii Aβ poate produce evenimente inflamatorii/oxidative secvențiale și excitotoxicitate, provocând neurodegenerare și tulburări cognitive [ 131 ]. În plus, studiile epidemiologice au arătat că utilizarea medicamentelor antiinflamatoare nesteroidiene (AINS) scade incidența AD [ 132 ]. S-a demonstrat că Aβ acționează ca un agent proinflamator, activând multe componente inflamatorii, iar SP, înconjurat de microglia și astrocite, coexistă cu citokine și chemokine [ 133] .]. Activarea microgliei indusă de Aβ este considerată a fi una dintre sursele majore ale răspunsului inflamator [ 134 ]. S-a raportat că melatonina atenuează răspunsurile microgliale și astrogliale induse de acid kainic, așa cum este determinat de detectarea imunohistochimică a izolectinei-B4 și a proteinei acide fibrilare gliale (GFAP), markerii specifici pentru microglia și respectiv astroglia [135 ] . Administrarea orală de melatonină a atenuat, de asemenea, citokinele proinflamatorii induse de Aβ, factorul nuclear κ B (NF- κ B) și oxidul nitric (NO) în creierul șobolanului [ 107 ].
Se raportează că melatonina a redus semnificativ răspunsul proinflamator, scăzând cu aproape 50% nivelurile induse de Aβ de citokine proinflamatorii, Interleukin-1-β (IL1-β), Interleukin-6 (IL6) și factorul de necroză tumorală-α (TNF). -α), in vivo [ 107 ]. Mai mult, activitatea de legare a ADN-ului NF- κB a fost inhibată de melatonină [ 136 , 137 ]. Mai recent, s-a demonstrat că melatonina reduce IL-6 indusă de NF- κ B într-o manieră dependentă de concentrație în feliile de creier tratate cu Aβ [ 138 ]. Administrarea melatoninei este, de asemenea, raportată că reduce învățarea indusă de Ap și afectarea memoriei la șobolani, împreună cu o scădere semnificativă a celulelor gliale pozitive care exprimă NF-IL-1β indusă de κ B în plus față de complementul 1q (C1q) în hipocamp [ 139 ].
Mergi la:
6. Concluzii
Melatonina este unul dintre cei mai puternici antioxidanți care acționează la diferite niveluri, iar nivelul melatoninei se reduce în timpul îmbătrânirii și la pacienții cu AD [ 24 , 40 , 140 – 142 ]. În plus, efectele sale antioxidante indirecte și efectele anti-amiloid se bazează pe sprijinul fazării circadiene și acțiunilor anti-excitotoxice adecvate [ 68 , 143] .]. Astfel, nu este surprinzător faptul că melatonina este protectoare în numeroase sisteme experimentale și a fost propusă ca tratament pentru AD. Studii recente de la șoareci transgenici APP au indicat că suplimentarea timpurie, pe termen lung, cu melatonină produce efecte anti-amiloid și antioxidante, dar nu se produce un astfel de efect atunci când tratamentul cu melatonină este inițiat după vârsta formării amiloidului [ 76 , 77 , 79 , 80 ] . Sunt necesare studii clinice extinse și studii cu modele transgenice pentru a confirma rolul melatoninei în stadiul patologic târziu al AD. Dacă melatonina nu are efect în stadiul târziu al AD, studiile asupra melatoninei ar trebui limitate la prevenirea AD, mai degrabă decât la tratament.
Pot apărea reacții adverse ale melatoninei, cum ar fi febră în prima zi de tratament cu melatonină, hiperkinezie sau plângeri de picioare agitate, menoragie, pigmentare la nivelul brațelor și picioarelor, dureri de cap și reacții abdominale, tromboză și somnolență [ 16 , 17 , 144 , 145 .]. În afară de aceste reacții adverse, aplicarea precoce și pe termen lung a melatoninei poate cel puțin încetini dezvoltarea AD. Pe lângă efectele pozitive în sistemele experimentale privind antagonismul deficitului colinergic, inflamația, fibrilogeneza și formarea încurcăturii, efectele de stimulare a somnului și suprimarea apusului sunt rezultate importante care justifică utilizarea melatoninei. Deși există dovezi care să postuleze melatonina ca un instrument util și terapeutic în MCI și AD, studii mai mari, dublu-orb, multicentrice, sunt necesare urgent pentru a explora și investiga în continuare potențialul și utilitatea melatoninei. Deoarece scăderea imunoreactivitatii MT2 și creșterea imunoreactivitatii MT1 au fost raportate în hipocampul pacienților cu AD [ 13 , 14] .], se așteaptă urgent regulatori specifici ai receptorilor de melatonină și noi derivați ai melatoninei.
Mergi la:
Mulțumiri
Această lucrare a fost susținută de granturi de la Fundația de Științe Naturale din China (30971204, 81271404).
Mergi la:
Conflict de interese
Autorii nu declară niciun conflict de interese.
Mergi la:
Referințe
1.
Lars MI, Jürgen G. Amyloid-β și tau—Un pas de deux toxic în boala Alzheimer. Nat. Pr. Neurosci. 2011; 12 :67–72. [ PubMed ] [ Google Scholar ]2.
Mustapic M., Popovic Hadzija M., Pavlovic M., Pavkovic P., Presecki P., Mrazovac D., Mimica N., Korolija M., Pivac N., Muck-Seler D. Boala Alzheimer și diabetul de tip 2 : Studiul de asociere al polimorfismelor în genele factorului de necroză tumorală-alfa și apolipoproteinei E. Metab. Brain Dis. 2012; 27 :507–512. [ PubMed ] [ Google Scholar ]3.
Ajala T., Rafi J., Wray R., Whitehead MW, Zaidi J. Poate exista o legătură între colestaza intrahepatică a sarcinii și hiperlipidemia combinată familială: un raport de caz. Cauzele J. 2009; 2 doi: 10.4076/1757-1626-2-8679. [ Articol gratuit PMC ] [ PubMed ] [ CrossRef ] [ Google Scholar ]4.
Leszek J., Sochocka M., Gasiorowski K. Factori vasculari și modificări epigenetice în patogeneza bolii Alzheimer. J. Neurol. Sci. 2012; 323 :25–32. [ PubMed ] [ Google Scholar ]5.
Rocchi A., Valensin D., Aldinucci C., Giani G., Barbucci R., Gaggelli E., Kozlowski H., Valensin G. Investigarea metabolomică RMN a astrocitelor a interacționat cu Abeta(42) sau cu complexele acestuia fie cu cuprul (II) sau zinc(II) J. Inorg. Biochim. 2012; 117 :326–333. [ PubMed ] [ Google Scholar ]6.
Rukhsana S., Butterfield DA Rolul stresului oxidativ în progresia bolii Alzheimer. J. Alzheimer Dis. 2010; 19 :341–353. [ PubMed ] [ Google Scholar ]7.
Wu YH, Swaab DF Glanda pineală umană și melatonina în îmbătrânire și boala Alzheimer. J. Pineal Res. 2005; 38 :145–152. [ PubMed ] [ Google Scholar ]8.
Wu YH, Feenstra MG, Zhou JN, Liu RY, Torano JS, van Kan HJ, Fischer DF, Ravid R., Swaab DF Modificări moleculare care stau la baza nivelurilor reduse de melatonina pineală în boala Alzheimer: Modificări în stadiile preclinice și clinice. J. Clin. Endocr. Metab. 2003; 88 :5898–5906. [ PubMed ] [ Google Scholar ]9.
Ferrari E., Arcaini A., Gornati R., Pelanconi L., Cravello L., Fioravanti M., Solerte SB, Magri F. Funcția pineală și hipofizo-adrenocorticală în îmbătrânirea fiziologică și în demența senilă. Exp. Gerontol. 2000; 35 :1239–1250. [ PubMed ] [ Google Scholar ]10.
Ohashi Y., Okamoto N., Uchida K., Iyo M., Mori N., Morita Y. Ritmul zilnic al nivelurilor serice de melatonina și efectul expunerii la lumină la pacienții cu demență de tip Alzheimer. Biol. Psihiatul. 1999; 45 :1646–1652. [ PubMed ] [ Google Scholar ]11.
Liu RY, Zhou JN, van Heerikhuize J., Hofman MA, Swaab DF Scăderea nivelului de melatonina în lichidul cefalorahidian postmortem în raport cu îmbătrânirea, boala Alzheimer și genotipul apolipoproteinei E-epsilon4/4. J. Clin. Endocr. Metab. 1999; 84 :323–327. [ PubMed ] [ Google Scholar ]12.
Zhou JN, Liu RY, Kamphorst W., Hofman MA, Swaab DF Modificările neuropatologice precoce ale Alzheimer la persoanele în vârstă sunt însoțite de scăderea nivelului de melatonină din lichidul cefalorahidian. J. Pineal Res. 2003; 35 :125–130. [ PubMed ] [ Google Scholar ]13.
Savaskan E., Olivieri G., Meier F., Brydon L., Jockers R., Ravid R., Wirz-Justice A., Muller-Spahn F. Imunoreactivitate crescută a receptorilor melatoninei 1a în hipocampul pacienților cu boala Alzheimer . J. Pineal Res. 2002; 32 :59–62. [ PubMed ] [ Google Scholar ]14.
Savaskan E., Ayoub MA, Ravid R., Angeloni D., Fraschini F., Meier F., Eckert A., Muller-Spahn F., Jockers R. Expresia redusă a receptorului de melatonină MT2 hipocampal în boala Alzheimer. J. Pineal Res. 2005; 38 :10–16. [ PubMed ] [ Google Scholar ]15.
Friedland RP, Luxenberg JS, Koss E. Un studiu cantitativ al calcificării intracraniene în demența de tip Alzheimer. Int. Psihogeriatr. 1990; 2 :36–43. [ PubMed ] [ Google Scholar ]16.
Wu YH, Fischer DF, Swaab DF Polimorfismul promotorului A în gena monoaminoxidază A este asociat cu activitatea MAOA pineală la pacienții cu boala Alzheimer. Brain Res. 2007; 1167 :13–19. [ PubMed ] [ Google Scholar ]17.
Cohen-Mansfield J., Garfinkel D., Lipson S. Melatonina pentru tratamentul apusului la persoanele în vârstă cu demență – Un studiu preliminar. Arc. Gerontol. Geriatr. 2000; 31 :65–76. [ PubMed ] [ Google Scholar ]18.
Brusco LI, Marquez M., Cardinali DP Tratamentul cu melatonina stabilizează simptomele cronobiologice și cognitive în boala Alzheimer. Neuro Endocrinol. Lett. 2000; 21 :39–42. [ PubMed ] [ Google Scholar ]19.
Brusco LI, Marquez M., Cardinali DP Gemeni monozigoți cu boala Alzheimer tratați cu melatonină: Raport de caz. J. Pineal Res. 1998; 25 :260–263. [ PubMed ] [ Google Scholar ]20.
Cardinali DP, Brusco LI, Perez Lloret S., Furio AM Melatonina în tulburările de somn și jet-lag. Neuro Endocrinol. Lett. 2002; 23 :9–13. [ PubMed ] [ Google Scholar ]21.
Cardinali DP, Brusco LI, Liberczuk C., Furio AM Utilizarea melatoninei în boala Alzheimer. Neuro Endocrinol. Lett. 2002; 23 :20–23. [ PubMed ] [ Google Scholar ]22.
Karasek M., Reiter RJ, Cardinali DP, Pawlikowski M. Future of melatonin as a therapeutic agent. Neuro Endocrinol. Lett. 2002; 23 :118–121. [ PubMed ] [ Google Scholar ]23.
Singer C., Tractenberg RE, Kaye J., Schafer K., Gamst A., Grundman M., Thomas R., Thal LJ Alzheimers cooperative, SA multicentric, studiu controlat cu placebo al melatoninei pentru tulburările de somn în boala Alzheimer . Dormi. 2003; 26 :893–901. [ Articol gratuit PMC ] [ PubMed ] [ Google Scholar ]24.
Ling ZQ, Tian Q., Wang L., Fu ZQ, Wang XC, Wang Q., Wang JZ Iluminarea constantă induce leziuni asemănătoare Alzheimer cu implicarea reticulului endoplasmatic și protecția melatoninei. J. Alzheimer Dis. 2009; 16 :287–300. [ PubMed ] [ Google Scholar ]25.
Selkoe DJ Biologia celulară a plierii greșite a proteinelor: exemplele bolilor Alzheimer și Parkinson. Nat. Cell Biol. 2004; 6 :1054–1061. [ PubMed ] [ Google Scholar ]26.
Brion JP, Anderton BH, Authelet M., Dayanandan R., Leroy K., Lovestone S., Octave JN, Pradier L., Touchet N., Tremp G. Neurofibrillary tangles and tau phosphorylation. Biochim. Soc. Symp. 2001; 67 :81–88. [ PubMed ] [ Google Scholar ]27.
Billingsley ML, Kincaid RL Fosforilarea și defosforilarea reglementate a proteinei tau: Efecte asupra interacțiunii microtubulilor, traficului intracelular și neurodegenerării. Biochim. J. 1997; 323 :577–591. [ Articol gratuit PMC ] [ PubMed ] [ Google Scholar ]28.
Braak E., Braak H., Mandelkow EM O secvență de modificări ale citoscheletului legate de formarea de încurcături neurofibrilare și fire de neuropil. Acta neuropathol. 1994; 87 :554–567. [ PubMed ] [ Google Scholar ]29.
Avila J., Perez M., Lucas JJ, Gomez-Ramos A., Santa Maria I., Moreno F., Smith M., Perry G., Hernandez F. Assembly in vitro of tau protein and its implications in Alzheimer’s boala. Curr. Alzheimer Res. 2004; 1 :97–101. [ PubMed ] [ Google Scholar ]30.
Sahara N., DeTure M., Ren Y., Ebrahim AS, Kang D., Knight J., Volbracht C., Pedersen JT, Dickson DW, Yen SH și colab. Caracteristicile speciilor Tau hiperfosforilate extractibile cu TBS: intermediari de agregare în creierul de șoarece rTg4510. J. Alzheimer Dis. 2013; 33 :249–263. [ Articol gratuit PMC ] [ PubMed ] [ Google Scholar ]31.
Lei P., Ayton S., Finkelstein DI, Spoerri L., Ciccotosto GD, Wright DK, Wong BX, Adlard PA, Cherny RA, Lam LQ, et al. Deficiența de Tau induce parkinsonismul cu demență prin afectarea exportului de fier mediat de APP. Nat. Med. 2012; 18 :291–295. [ PubMed ] [ Google Scholar ]32.
Khatoon S., Grundke-Iqbal I., Iqbal K. Nivelurile creierului de proteină tau asociată microtubulilor sunt crescute în boala Alzheimer: un test radioimuno-slot-blot pentru nanogramele proteinei. J. Neurochem. 1992; 59 :750–753. [ PubMed ] [ Google Scholar ]33.
Khatoon S., Grundke-Iqbal I., Iqbal K. Nivelurile de tau normal și anormal fosforilat în diferite compartimente celulare și regionale ale bolii Alzheimer și creierul de control. FEBS Lett. 1994; 351 :80–84. [ PubMed ] [ Google Scholar ]34.
Hanger DP, Betts JC, Loviny TL, Blackstock WP, Anderton BH Noi situsuri de fosforilare identificate în tau hiperfosforilat (filament elicoidal împerecheat-tau) din creierul bolii Alzheimer utilizând spectrometria de masă nanoelectrospray. J. Neurochem. 1998; 71 :2465–2476. [ PubMed ] [ Google Scholar ]35.
Hanger DP, Byers HL, Wray S., Leung KY, Saxton MJ, Seereeram A., Reynolds CH, Ward MA, Anderton BH Noile site-uri de fosforilare în tau din creierul Alzheimer susțin un rol pentru cazein kinaza 1 în patogeneza bolii. J. Biol. Chim. 2007; 282 :23645–23654. [ PubMed ] [ Google Scholar ]36.
Hasegawa M., Morishima-Kawashima M., Takio K., Suzuki M., Titani K., Ihara Y. Secvență de proteine și analize spectrometrice de masă ale tau în creierul bolii Alzheimer. J. Biol. Chim. 1992; 267 :17047–1754. [ PubMed ] [ Google Scholar ]37.
Morishima-Kawashima M., Hasegawa M., Takio K., Suzuki M., Yoshida H., Watanabe A., Titani K., Ihara Y. Hyperphosphorylation of tau in PHF. Neurobiol. Îmbătrânire. 1995; 16 :365–371. [ PubMed ] [ Google Scholar ]38.
Wang XF, Dong CF, Zhang J., Wan YZ, Li F., Huang YX, Han L., Shan B., Gao C., Han J. și colab. Proteina tau umană formează complex cu PrP și unii mutanți PrP legați de GSS și fCJD posedă activități de legare mai puternice cu tau in vitro . Mol. Celulă. Biochim. 2008; 310 :49–55. [ PubMed ] [ Google Scholar ]39.
Peng CX, Hu J., Liu D., Hong XP, Wu YY, Zhu LQ, Wang JZ Intervenția glicogen sintaza kinazei-3beta modificată de boală de către melatonină oprește patologia și deficitele de memorie la un model animal Alzheimer. Neurobiol. Îmbătrânire. 2013; 34 :1555–1563. [ PubMed ] [ Google Scholar ]40.
Peter TN, Irina A., Eileen HB, Constantin B., Heiko B., Nigel JC, Rudolph JC, Barbara JC, Peter D., Kelly DT și colab. Corelația modificărilor neuropatologice ale bolii Alzheimer cu statusul cognitiv: o revizuire a literaturii. J. neuropathol. Exp. Neurol. 2012; 71 :362–381. [ Articol gratuit PMC ] [ PubMed ] [ Google Scholar ]41.
Deng YQ, Xu GG, Duan P., Zhang Q., Wang JZ Efectele melatoninei asupra hiperfosforilării tau induse de wortmannin. Acta Pharmacol. Păcat. 2005; 26 :519–526. [ PubMed ] [ Google Scholar ]42.
Li XC, Wang ZF, Zhang JX, Wang Q., Wang JZ Efectul melatoninei asupra hiperfosforilării tau induse de calyculin A. EURO. J. Pharmacol. 2005; 510 :25–30. [ PubMed ] [ Google Scholar ]43.
Li SP, Deng YQ, Wang XC, Wang YP, Wang JZ Melatonina protejează celulele neuroblastomului SH-SY5Y de afectarea neurofilamentului indusă de caliculina A și neurotoxicitatea. J. Pineal Res. 2004; 36 :186–191. [ PubMed ] [ Google Scholar ]44.
Yang X., Yang Y., Fu Z., Li Y., Feng J., Luo J., Zhang Q., Wang Q., Tian Q. Melatonina ameliorează modificările patologice asemănătoare Alzheimer și afectarea reținerii memoriei spațiale induse de calyculin A. J. Psychopharmacol. 2011; 25 :1118–1125. [ PubMed ] [ Google Scholar ]45.
Wang YP, Li XT, Liu SJ, Zhou XW, Wang XC, Wang JZ Melatonina a ameliorat leziunile asemănătoare Alzheimer induse de acid okadaic. Acta Pharmacol. Păcat. 2004; 25 :276–280. [ PubMed ] [ Google Scholar ]46.
Liu SJ, Wang JZ Fosforilarea tau asemănătoare Alzheimer indusă de wortmannin in vivo și atenuarea acesteia de către melatonină. Acta Pharmacol. Păcat. 2002; 23 :183–187. [ PubMed ] [ Google Scholar ]47.
Wang DL, Ling ZQ, Cao FY, Zhu LQ, Wang JZ Melatonina atenuează supraactivarea proteinei kinazei A induse de izoproterenol și hiperfosforilarea tau în creierul șobolanului. J. Pineal Res. 2004; 37 :11–16. [ PubMed ] [ Google Scholar ]48.
Wang XC, Zhang J., Yu X., Han L., Zhou ZT, Zhang Y., Wang JZ Prevenirea hiperfosforilării tau indusă de izoproterenol de către melatonină la șobolan. Acta Pharmacol. Păcat. 2005; 57 :7–12. [ PubMed ] [ Google Scholar ]49.
Avila J. Agregarea Tau în polimeri fibrilari: Taupathies. FEBS Lett. 2000; 476 :89–92. [ PubMed ] [ Google Scholar ]50.
Gong CX, Liu F., Grundke-Iqbal I., Iqbal K. Modificări post-translaționale ale proteinei tau în boala Alzheimer. J. Transm. Neural. 2005; 112 :813–838. [ PubMed ] [ Google Scholar ]51.
Zhu LQ, Wang SH, Ling ZQ, Wang DL, Wang JZ Efectul inhibării biosintezei melatoninei asupra reținerii memoriei spațiale și fosforilării tau la șobolan. J. Pineal Res. 2004; 37 :71–77. [ PubMed ] [ Google Scholar ]52.
Reiter RJ, Acuna-Castroviejo D., Tan DX, Burkhardt S. Daune moleculare mediate de radicali liberi. Mecanisme pentru acțiunile protectoare ale melatoninei în sistemul nervos central. Ann. NY Acad. Sci. 2001; 939 :200–215. [ PubMed ] [ Google Scholar ]53.
Zhu X., Rottkamp CA, Boux H., Takeda A., Perry G., Smith MA Activarea kinazei p38 leagă fosforilarea tau, stresul oxidativ și evenimentele legate de ciclul celular în boala Alzheimer. J. Neuropat. Exp. Neur. 2000; 59 :880–888. [ PubMed ] [ Google Scholar ]54.
Gomez-Ramos A., Diaz-Nido J., Smith MA, Perry G., Avila J. Effect of the lipid peroxidation product acrolein on tau phosphorylation in neuronal cells. J. Neurosci. Res. 2003; 71 :863–870. [ PubMed ] [ Google Scholar ]55.
Lovell MA, Xiong S., Xie C., Davies P., Markesbery WR Inducerea tau hiperfosforilată în culturi primare de neuroni corticali de șobolan mediată de stresul oxidativ și glicogen sintaza kinaza-3. J. Alzheimer Dis. 2004; 6 :659–671. [ PubMed ] [ Google Scholar ]56.
Kenyon CJ Genetica îmbătrânirii. Natură. 2010; 464 :504–512. [ PubMed ] [ Google Scholar ]57.
Paradies G., Petrosillo G., Paradies V., Reiter RJ, Ruggiero FM Melatonina, cardiolipină și bioenergetica mitocondrială în sănătate și boală. J. Pineal Res. 2010; 48 :297–310. [ PubMed ] [ Google Scholar ]58.
Romero A., Egea J., Garcia AG, Lopez MG Efect neuroprotector sinergic al concentrațiilor scăzute combinate de galantamina și melatonină împotriva stresului oxidativ în celulele neuroblastomului SH-SY5Y. J. Pineal Res. 2010; 49 :141–148. [ PubMed ] [ Google Scholar ]59.
Hardeland R., Tan DX, Reiter RJ Kynuramines, metabolites of melatonin and other indoles: The resurrection of a near forgot class of biogenic amines. J. Pineal Res. 2009; 47 :109–126. [ PubMed ] [ Google Scholar ]60.
Jou MJ, Peng TI, Hsu LF, Jou SB, Reiter RJ, Yang CM, Chiao CC, Lin YF, Chen CC Vizualizarea nivelurilor multiple de protecție mitocondrială ale melatoninei împotriva tranziției de permeabilitate mediată de Ca(2+) mitocondrială și nu numai în astrocite ale creierului de șobolan. J. Pineal Res. 2010; 48 :20–38. [ PubMed ] [ Google Scholar ]61.
Hong Y., Palaksha KJ, Park K., Park S., Kim HD, Reiter RJ, Chang KT Melatonina plus terapie neuroreabilitativă bazată pe exerciții pentru leziuni ale măduvei spinării. J. Pineal Res. 2010; 49 :201–209. [ PubMed ] [ Google Scholar ]62.
Das A., McDowell M., Pava MJ, Smith JA, Reiter RJ, Woodward JJ, Varma AK, Ray SK, Banik NL Inhibarea apoptozei de către melatonină în motoneuronii VSC4.1 expuși la stres oxidativ, excitotoxicitate pe glutamat sau Toxicitatea TNF-alfa implică receptorii de melatonină de membrană. J. Pineal Res. 2010; 48 :157–169. [ Articol gratuit PMC ] [ PubMed ] [ Google Scholar ]63.
Schuster C., Williams LM, Morris A., Morgan PJ, Barrett P. Receptorul uman de melatonină MT1 stimulează producția de cAMP în celulele SH-SY5Y ale liniei celulare de neuroblastom uman printr-o cale de transducție a semnalului calciu-calmodulină. J. Neuroendocrinol. 2005; 17 :170–178. [ PubMed ] [ Google Scholar ]64.
Peschke E., Muhlbauer E., Musshoff U., Csernus VJ, Chankiewitz E., Peschke D. Receptorul (MT(1)) influența mediată a melatoninei asupra concentrației cAMP și secreției de insulină a celulelor de insulinom de șobolan INS-1. J. Pineal Res. 2002; 33 :63–71. [ PubMed ] [ Google Scholar ]65.
Witt-Enderby PA, MacKenzie RS, McKeon RM, Carroll EA, Bordt SL, Melan MA. Cell Motil. Citoscheletul. 2000; 46 :28–42. [ PubMed ] [ Google Scholar ]66.
Rivera-Bermudez MA, Gerdin MJ, Earnest DJ, Dubocovich ML Reglarea ritmicității bazale în activitatea protein kinazei C de către melatonina în celulele nucleului suprachiasmatic de șobolan imortalizate. Neurosci. Lett. 2003; 346 :37–40. [ PubMed ] [ Google Scholar ]67.
Benitez-King G., Rios A., Martinez A., Anton-Tay F. Inhibarea in vitro a activității kinazei II dependente de Ca2 + /calmodulină de către melatonină. Biochim. Biophys. Acta. 1996; 1290 :191–196. [ PubMed ] [ Google Scholar ]68.
Chen S., Xu Y., Xu B., Guo M., Zhang Z., Liu L., Ma H., Chen Z., Luo Y., Huang S., Chen L. CaMKII este implicat în cadmiu activarea căilor MAPK și mTOR care conduc la moartea celulelor neuronale. J. Neurochem. 2011; 119 :1108–1118. [ Articol gratuit PMC ] [ PubMed ] [ Google Scholar ]69.
Chan AS, Lai FP, Lo RK, Voyno-Yasenetskaya TA, Stanbridge EJ, Wong YH Receptorii melatoninei mt1 și MT2 stimulează kinaza N -terminală c-Jun prin proteinele G sensibile și insensibile la toxina pertussis. Celulă. Semnal. 2002; 14 :249–257. [ PubMed ] [ Google Scholar ]70.
Chen L., Xu B., Liu L., Luo Y., Yin J., Zhou H., Chen W., Shen T., Han X., Huang S. Peroxidul de hidrogen inhibă semnalizarea mTOR prin activarea AMPKalfa conducând la apoptoza celulelor neuronale. Lab Invest. 2010; 90 :762–773. [ Articol gratuit PMC ] [ PubMed ] [ Google Scholar ]71.
Chen L., Xu B., Liu L., Luo Y., Zhou H., Chen W., Shen T., Han X., Kontos CD, Huang S. Inducerea cadmiului a speciilor reactive de oxigen activează calea mTOR , ceea ce duce la moartea celulelor neuronale. Radic liber. Biol. Med. 2011; 50 :624–632. [ Articol gratuit PMC ] [ PubMed ] [ Google Scholar ]72.
Xu B., Chen S., Luo Y., Chen Z., Liu L., Zhou H., Chen W., Shen T., Han X., Chen L., Huang S. Semnalizarea calciului este implicată în apoptoza neuronală indusă de cadmiu prin inducerea speciilor reactive de oxigen și activarea rețelei MAPK/mTOR. Plus unu. 2011; 6 :e19052. [ Articol gratuit PMC ] [ PubMed ] [ Google Scholar ]73.
Selkoe DJ Boala Alzheimer este o insuficiență sinaptică. Ştiinţă. 2002; 298 :789–791. [ PubMed ] [ Google Scholar ]74.
Selkoe DJ Biologia celulară a proteinei precursoare de beta-amiloid și a presenilinei în boala Alzheimer. Trends Cell Biol. 1998; 8 :447–453. [ PubMed ] [ Google Scholar ]75.
Fisher A., Pittel Z., Haring R., Bar-Ner N., Kliger-Spatz M., Natan N., Egozi I., Sonego H., Marcovitch I., Brandeis R. M1 agoniştii muscarinici pot modula unele dintre semnele distinctive ale bolii Alzheimer: Implicații în terapia viitoare. J. Mol. Neurosci. 2003; 20 :349–356. [ PubMed ] [ Google Scholar ]76.
Lahiri DK Melatonina afectează metabolismul proteinei precursoare beta-amiloid în diferite tipuri de celule. J. Pineal Res. 1999; 26 :137–146. [ PubMed ] [ Google Scholar ]77.
Matsubara E., Bryant-Thomas T., Pacheco Quinto J., Henry TL, Poeggeler B., Herbert D., Cruz-Sanchez F., Chyan YJ, Smith MA, Perry G. și colab. Melatonina crește supraviețuirea și inhibă patologia oxidativă și amiloidă într-un model transgenic al bolii Alzheimer. J. Neurochem. 2003; 85 :1101–1108. [ PubMed ] [ Google Scholar ]78.
Lahiri DK, Chen D., Ge YW, Bondy SC, Sharman EH Suplimentarea alimentară cu melatonină reduce nivelurile de beta-peptide amiloide în cortexul cerebral murin. J. Pineal Res. 2004; 36 :224–231. [ PubMed ] [ Google Scholar ]79.
Song W., Lahiri DK Melatonina modifică metabolismul proteinei precursoare de beta-amiloid în linia celulară neuroendocrină PC12. J. Mol. Neurosci. 1997; 9 :75–92. [ PubMed ] [ Google Scholar ]80.
Zhang YC, Wang ZF, Wang Q., Wang YP, Wang JZ Melatonina atenuează inhibarea indusă de beta-amiloid a expresiei neurofilamentului. Acta Pharmacol. Păcat. 2004; 25 :447–451. [ PubMed ] [ Google Scholar ]81.
Olivieri G., Hess C., Savaskan E., Ly C., Meier F., Baysang G., Brockhaus M., Muller-Spahn F. Melatonina protejează celulele neuroblastomului SHSY5Y de stresul oxidativ indus de cobalt, neurotoxicitatea și creșterea secretia de beta-amiloid. J. Pineal Res. 2001; 31 :320–325. [ PubMed ] [ Google Scholar ]82.
Wang XC, Zhang YC, Chatterjie N., Grundke-Iqbal I., Iqbal K., Wang JZ Efectul melatoninei și melatonilvalpromidei asupra beta-amiloidului și neurofilamentelor în celulele N2a. Neurochema. Res. 2008; 33 :1138–1144. [ PubMed ] [ Google Scholar ]83.
Quinn J., Kulhanek D., Nowlin J., Jones R., Pratico D., Rokach J., Stackman R. Terapia cronică cu melatonină nu reușește să modifice încărcarea amiloidului sau deteriorarea oxidativă la șoarecii vechi Tg2576: Implicații pentru studiile clinice. Brain Res. 2005; 1037 :209–213. [ PubMed ] [ Google Scholar ]84.
Hsiao K., Chapman P., Nilsen S., Eckman C., Harigaya Y., Younkin S., Yang F., Cole G. Deficiențe de memorie corelative, creșterea Abeta și plăci de amiloid la șoarecii transgenici. Ştiinţă. 1996; 274 :99–102. [ PubMed ] [ Google Scholar ]85.
Su Y., Ryder J., Li B., Wu X., Fox N., Solenberg P., Brune K., Paul S., Zhou Y., Liu F. și colab. Litiul, un medicament comun pentru tratamentul tulburării bipolare, reglează procesarea proteinei precursoare de amiloid-beta. Biochimie. 2004; 43 :6899–6908. [ PubMed ] [ Google Scholar ]86.
Ryder J., Su Y., Liu F., Li B., Zhou Y., Ni B. Roluri divergente ale GSK-3 și CDK5 în procesarea APP. Biochim. Biophys. Res. comun. 2003; 312 :922–929. [ PubMed ] [ Google Scholar ]87.
Phiel CJ, Wilson CA, Lee VM, Klein PS GSK-3alpha reglează producția de peptide amiloid-beta ale bolii Alzheimer. Natură. 2003; 423 :435–439. [ PubMed ] [ Google Scholar ]88.
Donnelly PS, Caragounis A., Du T., Laughton KM, Volitakis I., Cherny RA, Sharples RA, Hill AF, Li QX, Masters CL și colab. Eliberarea intracelulară selectivă a ionilor de cupru și zinc din complecșii bis (thiosemicarbazonato) reduce nivelul peptidei amiloid-beta bolii Alzheimer. J. Biol. Chim. 2008; 283 :4568–4577. [ PubMed ] [ Google Scholar ]89.
White AR, Du T., Laughton KM, Volitakis I., Sharples RA, Xilinas ME, Hoke DE, Holsinger RM, Evin G., Cherny RA, et al. Degradarea beta-peptidei amiloid din boala Alzheimer prin reglarea în sus a activității metaloproteazei dependentă de metal. J. Biol. Chim. 2006; 281 :17670–17680. [ PubMed ] [ Google Scholar ]90.
Tesco G., Tanzi RE GSK-3 beta formează un complex tetrameric cu PS1-CTF/NTF endogen și beta-catenina. Efectele mutațiilor legate de D257/D385A și FAD. Ann. NY Acad. Sci. 2000; 920 :227–232. [ PubMed ] [ Google Scholar ]91.
Takashima A., Murayama M., Murayama O., Kohno T., Honda T., Yasutake K., Nihonmatsu N., Mercken M., Yamaguchi H., Sugihara S., et al. Presenilina 1 se asociază cu glicogen sintaza kinaza-3beta și substratul său tau. Proc. Natl. Acad. Sci. STATELE UNITE ALE AMERICII. 1998; 95 :9637–9641. [ Articol gratuit PMC ] [ PubMed ] [ Google Scholar ]92.
Simmons LK, May PC, Tomaselli KJ, Rydel RE, Fuson KS, Brigham EF, Wright S., Lieberburg I., Becker GW, Brems DN și colab. Structura secundară a peptidei beta-amiloid se corelează cu activitatea neurotoxică in vitro . Mol. Pharmacol. 1994; 45 :373–379. [ PubMed ] [ Google Scholar ]93.
Soto C., Castano EM Conformația peptidei beta Alzheimer determină viteza de formare a amiloidului și rezistența acestuia la proteoliză. Biochim. J. 1996; 314 :701–707. [ Articol gratuit PMC ] [ PubMed ] [ Google Scholar ]94.
Poeggeler B., Miravalle L., Zagorski MG, Wisniewski T., Chyan YJ, Zhang Y., Shao H., Bryant-Thomas T., Vidal R., Frangione B. și colab. Melatonina inversează activitatea profibrilogenă a apolipoproteinei E4 asupra peptidei amiloid Abeta Alzheimer. Biochimie. 2001; 40 :14995–5001. [ PubMed ] [ Google Scholar ]95.
Skribanek Z., Balaspiri L., Mak M. Interacțiunea dintre amiloid-beta-peptidă sintetică (1–40) și inhibitorii săi de agregare studiați prin spectrometrie de masă cu ionizare electrospray. J. Spectrom de masă. 2001; 36 :1226–1229. [ PubMed ] [ Google Scholar ]96.
Pappolla M., Bozner P., Soto C., Shao H., Robakis NK, Zagorski M., Frangione B., Ghiso J. Inhibirea beta-fibrilogenezei Alzheimer de către melatonină. J. Biol. Chim. 1998; 273 :7185–7188. [ PubMed ] [ Google Scholar ]97.
Huang TH, Fraser PE, Chakrabartty A. Fibrillogenesis of Alzheimer’s Abeta peptides studied by fluorescence energy transfer. J. Mol. Biol. 1997; 269 :214–224. [ PubMed ] [ Google Scholar ]98.
Fraser PE, Nguyen JT, Surewicz WK, Kirschner DA tranziții structurale dependente de pH ale peptidelor amiloide Alzheimer. Biophys. J. 1991; 60 :1190–1201. [ Articol gratuit PMC ] [ PubMed ] [ Google Scholar ]99.
Masilamoni JG, Jesudason EP, Dhandayuthapani S., Ashok BS, Vignesh S., Jebaraj WC, Paul SF, Jayakumar R. Rolul neuroprotector al melatoninei împotriva șoarecilor injectați cu peptida beta amiloid. Radic liber. Res. 2008; 42 :661–673. [ PubMed ] [ Google Scholar ]100.
Pacchierotti C., Iapichino S., Bossini L., Pieraccini F., Castrogiovanni P. Melatonin in psychiatric disorders: A review on the melatonin involvement in psychiatry. Față. Neuroendocrină. 2001; 22 :18–32. [ PubMed ] [ Google Scholar ]101.
Yuan H., Pang SF [125I] Situri de legare a iodomelatoninei în creierul porumbeilor: caracteristici de legare, distribuție regională și variație diurnă. J. Endocrinol. 1991; 128 :475–482. [ PubMed ] [ Google Scholar ]102.
Bieschke J., Zhang Q., Powers ET, Lerner RA, Kelly JW Metaboliții oxidativi accelerează amiloidogeneza Alzheimer printr-un mecanism în două etape, eliminând cerința de nucleare. Biochimie. 2005; 44 :4977–4983. [ PubMed ] [ Google Scholar ]103.
Feng Z., Zhang JT Efectul protector al melatoninei asupra apoptozei induse de beta-amiloid în celulele C6 de astrogliom de șobolan și mecanismul acestuia. Radic liber. Biol. Med. 2004; 37 :1790–1801. [ PubMed ] [ Google Scholar ]104.
Zatta P., Tognon G., Carampin P. Melatonina previne formarea radicalilor liberi datorită interacțiunii dintre peptidele beta-amiloide și metalioni [Al(III), Zn(II), Cu(II), Mn(II), Fe(II)] J. Pineal Res. 2003; 35 :98–103. [ PubMed ] [ Google Scholar ]105.
Feng Z., Chang Y., Cheng Y., Zhang BL, Qu ZW, Qin C., Zhang JT Melatonina atenuează deficitele comportamentale asociate cu apoptoza și disfuncția sistemului colinergic în modelul de șoarece transgenic APP 695 al bolii Alzheimer. J. Pineal Res. 2004; 37 :129–136. [ PubMed ] [ Google Scholar ]106.
Shen YX, Xu SY, Wei W., Sun XX, Liu LH, Yang J., Dong C. Efectele protectoare ale melatoninei de deteriorarea oxidativă indusă de beta-peptida amiloid 25–35 la șobolanii de vârstă mijlocie. J. Pineal Res. 2002; 32 :85–89. [ PubMed ] [ Google Scholar ]107.
Rosales-Corral S., Tan DX, Reiter RJ, Valdivia-Velazquez M., Martinez-Barboza G., Acosta-Martinez JP, Ortiz GG Melatonina administrată oral reduce stresul oxidativ și citokinele proinflamatorii induse de peptida beta-amiloid la șobolan creier: un studiu comparativ, in vivo versus vitamina C și E. J. Pineal Res. 2003; 35 :80–84. [ PubMed ] [ Google Scholar ]108.
Slats D., Claassen JA, Verbeek MM, Overeem S. Interacțiuni reciproce între somn, ritmuri circadiene și boala Alzheimer: Focus pe rolul hipocretinei și melatoninei. Imbatranire Res. Rev. 2013; 12 :188–200. [ PubMed ] [ Google Scholar ]109.
Rothman SM, Mattson MP Tulburări de somn în bolile Alzheimer și Parkinson. Neuromolecular Med. 2012; 14 :194–204. [ Articol gratuit PMC ] [ PubMed ] [ Google Scholar ]110.
Cecon E., Markus RP Relevanța acțiunilor cronobiologice și non-cronobiologice ale melatoninei pentru creșterea eficacității terapeutice în tulburările neurodegenerative. Brevetul recent. Endocr. Metab. Drug Imun Discov. 2011; 5 :91–99. [ PubMed ] [ Google Scholar ]111.
Kang JE, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, Fujiki N., Nishino S., Holtzman DM Dinamica beta-amiloid este reglată de orexină și de ciclul somn-veghe. Ştiinţă. 2009; 326 :1005–1007. [ Articol gratuit PMC ] [ PubMed ] [ Google Scholar ]112.
Struble RG, Cork LC, Whitehouse PJ, Price DL Inervația colinergică în plăcile nevrite. Ştiinţă. 1982; 216 :413–415. [ PubMed ] [ Google Scholar ]113.
Coyle JT, Price DL, DeLong MR Boala Alzheimer: O tulburare a inervației colinergice corticale. Ştiinţă. 1983; 219 :1184–1190. [ PubMed ] [ Google Scholar ]114.
Rasool CG, Svendsen CN, Selkoe DJ Degenerarea neurofibrilară a neuronilor colinergici și noncolinergici ai creierului anterior bazal în boala Alzheimer. Ann. Neurol. 1986; 20 :482–488. [ PubMed ] [ Google Scholar ]115.
Samuel W., Masliah E., Hill LR, Butters N., Terry R. Hipocampal connectivity and Alzheimer’s dementa: Effects of synapse loss and tangle frequency in a two-component model. Neurologie. 1994; 44 :2081–2088. [ PubMed ] [ Google Scholar ]116.
Davis KL, Mohs RC, Marin D., Purohit DP, Perl DP, Lantz M., Austin G., Haroutunian V. Markeri colinergici la pacienții vârstnici cu semne precoce ale bolii Alzheimer. J. Am. Med. conf. univ. 1999; 281 :1401–1406. [ PubMed ] [ Google Scholar ]117.
Rinne JO, Laine M., Hiltunen J., Erkinjuntti T. Luarea deciziilor semantice în AD probabilă timpurie: Un studiu de activare PET. Cogn. Brain Res. 2003; 18 :89–96. [ PubMed ] [ Google Scholar ]118.
Terry AV, Jr, Buccafusco JJ Ipoteza colinergică a vârstei și a deficitelor cognitive legate de boala Alzheimer: provocări recente și implicațiile lor pentru dezvoltarea de noi medicamente. J. Pharmacol. Exp. Acolo. 2003; 306 :821–827. [ PubMed ] [ Google Scholar ]119.
Spencer JP, Middleton LJ, Davies CH Investigarea eficacității inhibitorului de acetilcolinesterază, a donepezilului și a noilor agenți procognitivi pentru a induce oscilații gamma în feliile de hipocamp de șobolan. Neurofarmacologie. 2010; 59 :437–443. [ PubMed ] [ Google Scholar ]120.
Guermonprez L., Ducrocq C., Gaudry-Talarmain YM Inhibarea sintezei acetilcolinei și nitrarea tirozinei induse de peroxinitrit sunt prevenite diferențial de antioxidanți. Mol. Pharmacol. 2001; 60 :838–846. [ PubMed ] [ Google Scholar ]121.
Feng Z., Cheng Y., Zhang JT Efectele pe termen lung ale melatoninei sau 17 beta-estradiol asupra îmbunătățirii performanței memoriei spațiale la șobolani adulți cu deficiențe cognitive, ovariectomizate. J. Pineal Res. 2004; 37 :198–206. [ PubMed ] [ Google Scholar ]122.
Tang F., Nag S., Shiu SY, Pang SF Efectele melatoninei și extractului de Ginkgo biloba asupra pierderii memoriei și activităților colin acetiltransferazei în creierul șobolanilor infuzați intracerebroventricular cu beta-amiloid 1-40. Life Sci. 2002; 71 :2625–2631. [ PubMed ] [ Google Scholar ]123.
Agrawal R., Tyagi E., Shukla R., Nath C. Un studiu al receptorilor de insulină din creier, activitatea AChE și stresul oxidativ în modelul de șobolan al demenței induse de ICV STZ. Neurofarmacologie. 2009; 56 :779–787. [ PubMed ] [ Google Scholar ]124.
Hansen RA, Gartlehner G., Webb AP, Morgan LC, Moore CG, Jonas DE Eficacitatea și siguranța donepezilului, galantamina și rivastigminei pentru tratamentul bolii Alzheimer: o revizuire sistematică și meta-analiză. Clin. Interv. Îmbătrânire. 2008; 3 :211–225. [ Articol gratuit PMC ] [ PubMed ] [ Google Scholar ]125.
Cardinali DP, Furio AM, Brusco LI Aspecte clinice ale intervenției melatoninei în progresia bolii Alzheimer. Curr. Neurofarmacol. 2010; 8 :218–227. [ Articol gratuit PMC ] [ PubMed ] [ Google Scholar ]126.
Fernandez-Bachiller MI, Perez C., Campillo NE, Paez JA, Gonzalez-Munoz GC, Usan P., Garcia-Palomero E., Lopez MG, Villarroya M., Garcia AG, et al. Hibrizii tacrină-melatonină ca agenți multifuncționali pentru boala Alzheimer, cu proprietăți colinergice, antioxidante și neuroprotectoare. ChemMedChem. 2009; 4 :828–841. [ PubMed ] [ Google Scholar ]127.
Spuch C., Antequera D., Isabel Fernandez-Bachiller M., Isabel Rodriguez-Franco M., Carro E. Un nou hibrid tacrină-melatonină reduce sarcina de amiloid și deficitele comportamentale într-un model de șoarece al bolii Alzheimer. Neurotox. Res. 2010; 17 :421–431. [ PubMed ] [ Google Scholar ]128.
Arends YM, Duyckaerts C., Rozemuller JM, Eikelenboom P., Hauw JJ Microglia, amiloid și demență în boala Alzheimer. Un studiu corelativ. Neurobiol. Îmbătrânire. 2000; 21 :39–47. [ PubMed ] [ Google Scholar ]129.
Combadiere C., Feumi C., Raoul W., Keller N., Rodero M., Pezard A., Lavalette S., Houssier M., Jonet L., Picard E., et al. Acumularea de celule microglia subretiniene dependentă de CX3CR1 este asociată cu caracteristicile cardinale ale degenerescenței maculare legate de vârstă. J. Clin. Investi. 2007; 117 :2920–2928. [ Articol gratuit PMC ] [ PubMed ] [ Google Scholar ]130.
Streit WJ, Mrak RE, Griffin WS Microglia și neuroinflamație: O perspectivă patologică. J. Neuroinflamm. 2004; 1 doi: 10.1186/1742-2094-1-14. [ Articol gratuit PMC ] [ PubMed ] [ CrossRef ] [ Google Scholar ]131.
Hardy JA, Higgins GA Boala Alzheimer: Ipoteza cascadei amiloide. Ştiinţă. 1992; 256 :184–185. [ PubMed ] [ Google Scholar ]132.
Stuchbury G., Munch G. Inflamația asociată cu Alzheimer, potențiale ținte de droguri și terapii viitoare. J. Neural. Transm. 2005; 112 :429–453. [ PubMed ] [ Google Scholar ]133.
Tuppo EE, Arias HR Rolul inflamației în boala Alzheimer. Int. J. Biochim. Celula B. 2005; 37 :289–305. [ PubMed ] [ Google Scholar ]134.
Park SY, Jin ML, Kim YH, Kim Y., Lee SJ Efecte antiinflamatorii ale turmeronului aromatic prin blocarea căilor de semnalizare NF-kappaB, JNK și p38 MAPK în microglia stimulată de beta-amiloid. Int. Imunofarmacol. 2012; 14 :13–20. [ PubMed ] [ Google Scholar ]135.
Chung SY, Han SH Melatonina atenuează neurodegenerarea hipocampului indusă de acid kainic și stresul oxidativ prin inhibarea microgliale. J. Pineal Res. 2003; 34 :95–102. [ PubMed ] [ Google Scholar ]136.
Mohan N., Sadeghi K., Reiter RJ, Meltz ML Neurohormonul melatonina inhibă citokinele, mitogenele și radiațiile ionizante induse de NF-kappa B. Biochem. Mol. Biol. Int. 1995; 37 :1063–1070. [ PubMed ] [ Google Scholar ]137.
Chuang JI, Mohan N., Meltz ML, Reiter RJ Efectul melatoninei asupra activității de legare a ADN-ului NF-kappa-B în splina șobolanului. Cell Biol. Int. 1996; 20 :687–692. [ PubMed ] [ Google Scholar ]138.
Lau WW, Ng JK, Lee MM, Chan AS, Wong YH Interleukin-6 semnalizarea autocrină mediază fosforilarea STAT3 Tyr(705) indusă de receptorul melatoninei MT(1/2). J. Pineal Res. 2012; 52 :477–489. [ PubMed ] [ Google Scholar ]139.
Shen Y., Zhang G., Liu L., Xu S. Efectele supresive ale melatoninei asupra activării gliale induse de beta amiloid în hipocampul șobolanului. Arc. Med. Res. 2007; 38 :284–290. [ PubMed ] [ Google Scholar ]140.
Grases F., Costa-Bauza A., Prieto RM Un rol potențial al inhibitorilor de cristalizare în tratamentul bolii Alzheimer. Med. Ipoteza. 2010; 74 :118–119. [ PubMed ] [ Google Scholar ]141.
Boala Alzheimer Wollen KA: Avantajele și dezavantajele terapiilor farmaceutice, nutriționale, botanice și stimulatoare, cu o discuție despre strategiile de tratament din perspectiva pacienților și a practicienilor. J. Clin. Acolo. 2010; 15 :223–244. [ PubMed ] [ Google Scholar ]142.
Dowling GA, Burr RL, van Someren EJ, Hubbard EM, Luxenberg JS, Mastick J., Cooper BA Melatonina și tratamentul cu lumină puternică pentru perturbarea activității de repaus la pacienții instituționalizați cu boala Alzheimer. J. Am. Geriatr. Soc. 2008; 56 :239–246. [ Articol gratuit PMC ] [ PubMed ] [ Google Scholar ]143.
Pappolla MA, Chyan YJ, Poeggeler B., Frangione B., Wilson G., Ghiso J., Reiter RJ O evaluare a proprietăților antioxidante și antiamiloidogene ale melatoninei: Implicații pentru boala Alzheimer. J. Neural. Transm. 2000; 107 :203–231. [ PubMed ] [ Google Scholar ]144.
Nagtegaal J., Smits M., Van Der Meer Y., Fischer-Steenvoorden M. Melatonin: Un studiu asupra reacțiilor adverse suspectate la medicamente. Somn-Trezire Res. Netherl. 1996; 7 :115–118. [ Google Scholar ]145.
Avery D., Lenz M., Landis C. Linii directoare pentru prescrierea melatoninei. Ann. Med. 1998; 30 :122–130. [ PubMed ] [ Google Scholar ]
Articole din
Jurnalul Internațional de Științe Moleculare sunt furnizate aici prin amabilitatea
Multidisciplinary Digital Publishing Institute (MDPI)
SPUNE SI ALTORA (click pe butoane):

























{kind=link}
{kind=link}
{kind=link}
{kind=link}